CRISPR-Cas9の導入方法(プロトコール)と応用例とは?

CRISPR-Cas9は世界中の研究室で使われており、この記事の読者の中には「そろそろ自分の研究でもCRISPR-Cas9を使って実験をしたい」と考えている人がいるかもしれません。
そこで本記事では、CRISPR-Cas9を使った具体的なプロトコールや、遺伝子の発現抑制・過剰発現、スクリーニングへの応用例を紹介します。
培養細胞での一般的なCRISPR-Cas9導入方法(プロトコール)
一般的な培養細胞を用いたCRISPR-Cas9実験のワークフローの概要を図1に示します。
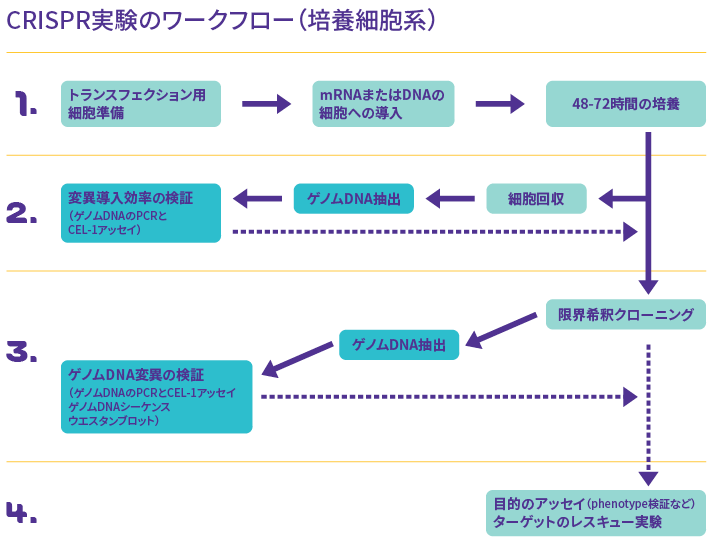
図1 CRISPR-Cas9実験のワークフロー(培養細胞系)
ステップ1 CRISPR-Cas9ツールの細胞導入
CRISPR-Cas9ツールは、DNAを切断するCas9ヌクレアーゼと、切断領域を認識してCas9と結合するガイドRNA(gRNA)から構成されています。
ツールを細胞に導入する方法には、ツールをコードするプラスミドDNAをトランスフェクションするプラスミド法と、ガイドRNA(gRNA)とCas9タンパク質の複合体(RNP: ribonucleoprotein)を試験管内で調整してから細胞に導入するRNP法の2つがあります。
<プラスミド法>
ベクターの種類として、gRNAとCas9が別々のベクターにコードされている個別プラスミド型と、gRNAとCas9を1つのプラスミドに収めた単一ベクター型(all-in-One)があります。
all-in-Oneシステムは、gRNAとCas9を効率よく同時発現できます。一方で個別プラスミド型は、変異導入効率が悪い場合やオフターゲット作用が増加する場合などに、gRNAやCas9プラスミド量を柔軟に変更して検討することが可能です。
導入時には条件検討が必要です。プラスミドDNAの場合、最初に推奨される使用量と濃度は、50万〜100万個の細胞に対して以下のとおりです。
- 1.0 μg/μLに調整したCas9プラスミドを5 μL以下
- 1.0 μg/μLに調整したgRNAプラスミドを5 μL以下
- プラスミド量は合計2〜8μg(all-in-Oneのようなsingle plasmidの場合)
プラスミドのフォーマットであれば、エレクトロポレーション法やリポソーム法など、ほとんどのトランスフェクション法を用いることが可能です。トランスフェクション試薬には、XTG360-RO Roche X-tremeGENE™ 360 Transfection Reagentなどがあります。
一般的なトランスフェクションと同様に、不死化細胞株にはほとんどのトランスフェクション法を利用できるのに対し、T細胞などの初代細胞ではトランスフェクションがより難しくなります。
その場合は、より効率のよい試薬を検討するか、ウイルスベクターによる導入を検討します。
<RNP法>
RNP法ではDNAプラスミドを使用せず、gRNAとCas9タンパク質の複合体を試験管内で調整してから細胞に導入します(図2)。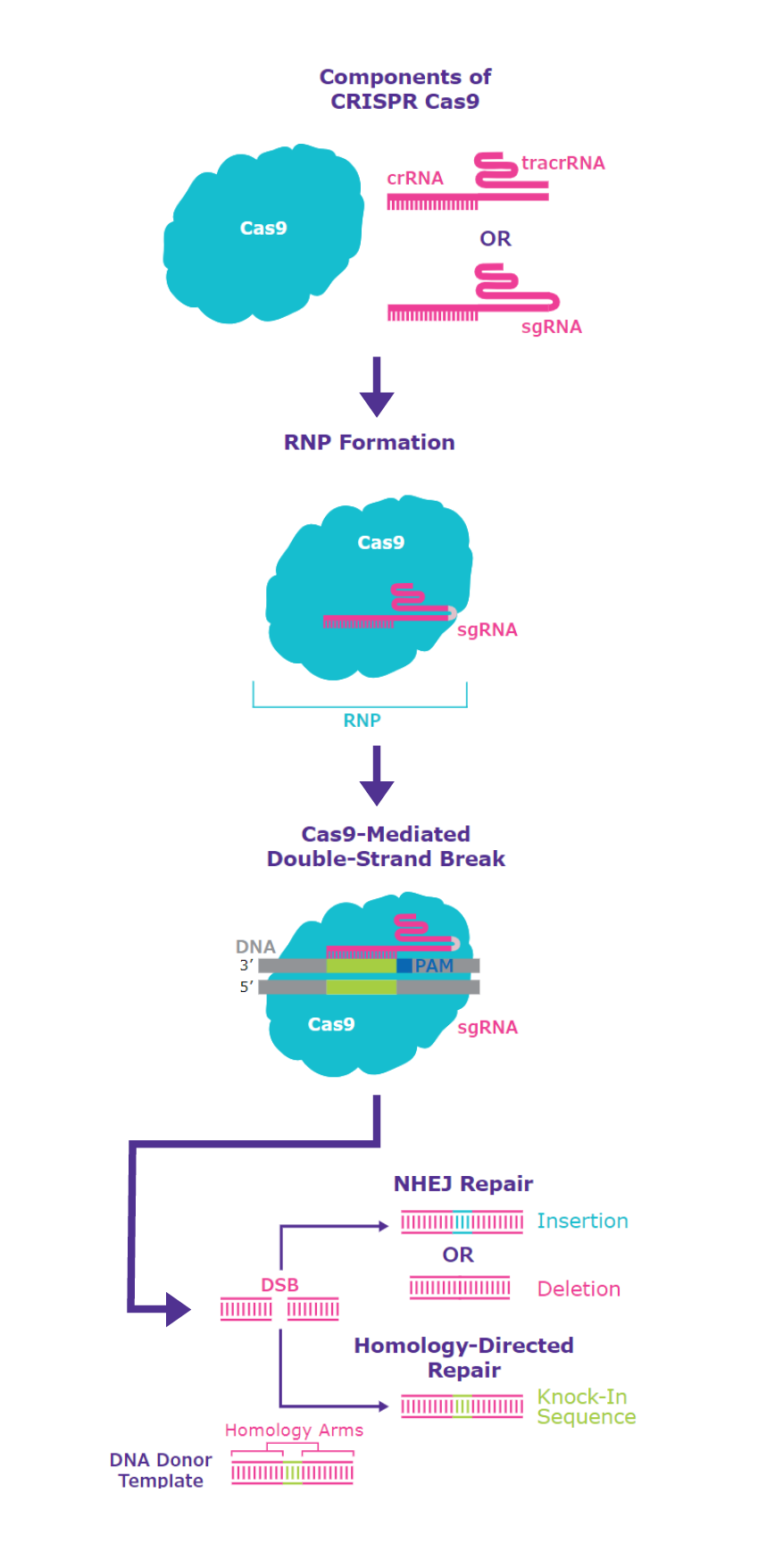
図2 RNP法の概要
プレデザイン品の無い生物種(ヒト・マウス以外)や、多数のgRNAを用いる場合など、コストを低く抑えたい場合に推奨されます。
また、転写と翻訳のステップが省かれ、Cas9はトランスフェクション後迅速に分解されるため、オフターゲット効果(オフターゲット作用)を抑えることができ、遺伝子ノックアウトの効率は70〜80%に達するとされています*1。
RNP法では、トランスフェクション試薬としてTransIT-CRISPRをおすすめします。TransIT-CRISPRは非リポソームポリマートランスフェクション試薬で、初代細胞を含むさまざまな細胞種に対して、迅速かつ高効率にRNPをトランスフェクションできます。
TransIT-CRISPRトランスフェクション試薬についてはこちら(英語版PDF)
具体的なRNP法のプロトコールは、こちらのページをご覧ください。(注:現在、合成gRNAは国内では販売しておりません)
ステップ2 変異導入効率の検証
CRISPR-Cas9システムを導入して48〜72時間培養したのち、変異導入効率を検証します。
この検証でよく使われるのは、CEL-1アッセイです。CEL-1は、ミスマッチ部分のDNA2本鎖を切断する酵素です。まず、CRISPRターゲット領域周辺の配列をPCRで増幅します。
ゲノム編集による変異が入っていれば、PCR産物を熱変性、アニーリングするとミスマッチ2本鎖が生じます。このミスマッチ変異をCEL-1酵素で切断し、電気泳動によって確認するのがCEL-1アッセイです(図3)。
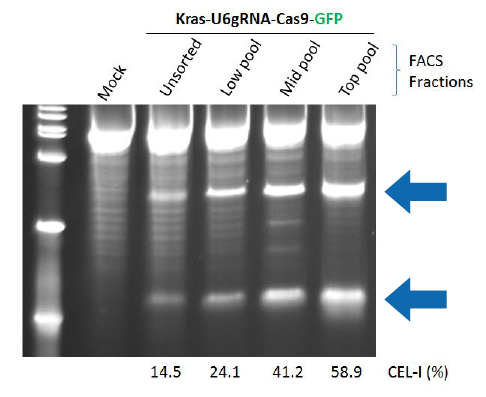
図3ヒトK562細胞にKRAS遺伝子領域を標的としたCRISPR-Cas9システムを導入後、Cas9-GFPの蛍光でFACSソーティングし、CEL-1アッセイを行った結果。GFPの蛍光強度が強い細胞集団ほど短い2本のバンドが濃く表れており、CEL-1による切断が起きている(すなわち変異が導入された)ことを意味する。
他に、リアルタイムPCRを用いて1塩基置換や挿入・欠損を判別できる高解像度融解曲曲線分析(HRM: high resolution melting)という方法もあります。
変異導入効率が悪い場合には、導入プラスミドの量を検討します。またトランスフェクション効率が悪いと判断された場合には、より効率よく導入できるトランスフェクション試薬への変更や、感染効率の高いウイルスフォーマットを検討します。
ステップ3 細胞のクローニングとゲノムDNA変異の検証
CEL-1アッセイによって変異導入の効率が十分なレベルであると判断された場合には、限界希釈クローニングやソーティング等を行い、シングルコロニー単位で変異を検証します。
このときにはCEL-1アッセイだけでなく、DNAシーケンスを行います。あるいは標的遺伝子がコードするタンパク質の発現量を調べるウエスタンブロットで検証する方法もあります。
ステップ4 変異細胞を用いた各種実験
ゲノム編集の成功が確認できたら、その細胞を用いて目的の実験を行えるようになります。
その他のCRISPR-Cas9導入方法
iPSCプロトコール
人工多能性幹細胞(iPSC: induced pluripotent stem cells)にCRISPR-Cas9を導入する際には、高品質なiPSCを用意することが重要です。
培養したiPSCの90%以上が多能性を保ち(OCT-4やNANOGなどの多能性マーカーで確認)、かつ何らかの細胞に分化してないことを確認することが、CRISPR-Cas9実験の成功のポイントとなります。
トランスジェニックマウス作製
gRNAを2種類用意すれば2重ノックアウトマウスを作製でき、ドナーテンプレートDNAを導入すればノックインマウスを作製できます。
トランスジェニックマウス作製の際には、適切なマウス系統を選択することが重要です。
スクリーニング
CRISPR-Cas9は簡便に遺伝子をノックアウトできることから、ヒトおよびマウスの全ゲノム遺伝子ライブラリーによるスクリーニングにも活用されています。ライブラリーにはプール型とアレイ型の2つがあります。
プール型ライブラリーはロボット工学や特殊な機器を使用せずに、ベンチトップでヒトとマウスのゲノム全体をスクリーニングすることができます。プール型の結果の判定には次世代シーケンサー(NGS)が必要です。
一方でアレイ型ライブラリーでは、gRNAがハイスループットスクリーニングに適した96ウェルフォーマットで提供されます。結果の判定に次世代シーケンサー(NGS)が不要で、ハイスループット顕微鏡法や、蛍光または発光検出による表現型変化で判定します。
弊社ではFeng Zhang博士およびMIT / Broad Instituteと提携し、改良されたプール型GeCKOv2ライブラリーや、Wellcome Sanger Instituteと共同で作成したアレイ型ライブラリーを提供しています。
プール型ライブラリーの詳細についてはこちら
アレイ型CRISPRライブラリーについてはこちら
CRISPR screeningのカスタマイズなど、詳細についてはご相談フォームからお問い合わせください。オンライン面談でもご説明可能です。
CRISPR-Cas9の応用例
CRISPRi、CRISPRa
CRISPR-Cas9はノックアウトまたはノックインを可能にする技術ですが、Cas9を改変して発現抑制または過剰発現することもできるようになりました。
Cas9の2つのヌクレアーゼドメインに点変異を導入したdCas9はDNAを切断できませんが、gRNAと協調して標的配列に結合する機能を有しています。dCas9に転写抑制因子であるKRABドメインを融合させると、標的遺伝子の転写を抑制し、ノックダウンが可能となります*2,3。これがCRISPR干渉(CRISPRi)です(図4)。
スクリーニング用に、がん・アポトーシス、膜タンパク質など分野に特化したライブラリーもあります(国内販売準備中)。
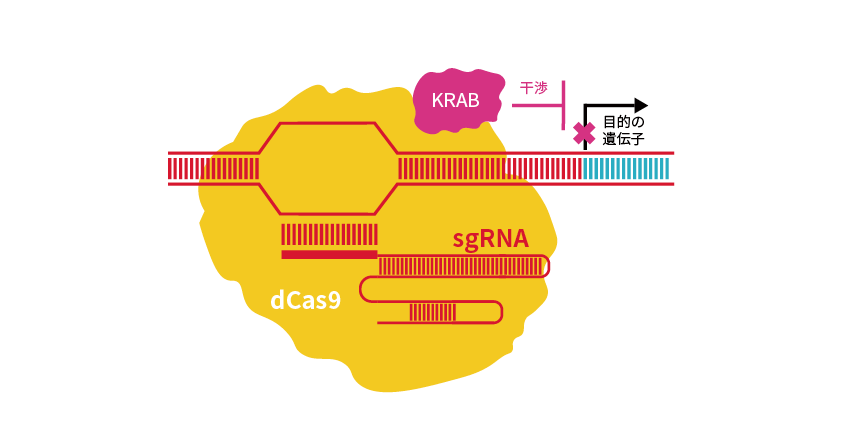
図4 KRABドメインを利用したCRISPRi
一方、dCas9にウイルス由来の転写活性化因子であるVP64を融合させ、過剰発現を可能にしたのがCRISPR activation(CRISPRa)です*4,5。
gRNAにはMS2 RNAアプタマーが含まれており、MS2-p65-HSF1活性化ドメインと組み合わせることで、VP64、p65、HSF1が相乗的に遺伝子発現を活性化します(図5)。ヒト、マウスに対する全ゲノムライブラリーもあります。
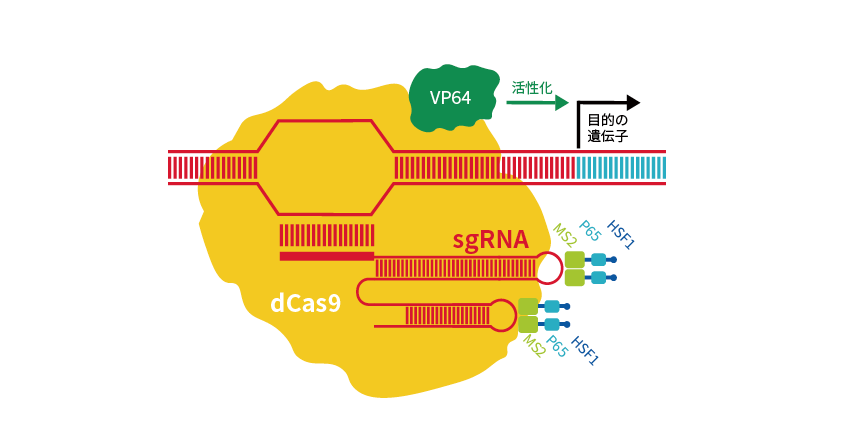 図5 VP64、p65、HSF1を利用したCRISPRa
図5 VP64、p65、HSF1を利用したCRISPRa
それぞれの詳細はこちらのページをご覧ください。
また、dCas9にシチジンデアミナーゼなど融合させて特定の1塩基を別の塩基に置換できる塩基編集*6や、 ヒストンアセチルトランスフェラーゼを融合させてエピジェネティック修飾を可能にするエピゲノム編集*7という技術もあります。
DNA鎖の1本だけを切断できるようにし、逆転写酵素を利用して新しいDNA配列を挿入するプライム編集という方法も開発されています*8。
今後はCRISPRをベースとした、より多様化したゲノム編集技術が利用可能になっていくでしょう。
シングルセル解析
プール型CRISPRスクリーニングを利用することで、シングルセルレベルでの解析も可能です。
数十〜数千のgRNAを単一のプールとして調製して培養細胞に導入し、各細胞に対して1種類のgRNAが導入されるようにします。解析にはNGSとデータのデコンボリューションを必要としますが、ゲノムワイドなトランスクリプトームプロファイルを中心とした高度な情報を得ることができます。
弊社では10x Genomics社との提携により、シングルセルレベルでの解析を可能にしたプール型CRISPRスクリーニング用製品を用意しています。詳細はこちらのページをご覧ください。
まとめ
この記事では、CRISPR-Cas9を用いた具体的なプロトコールと応用例を紹介しました。CRISPR-Cas9技術の発展は目覚ましく、新しい活用例が次々と登場しています。
メルクでは、CRISPR-Cas9についてまとめた無料の日本語ガイドとカタログを用意しております。CRISPR-Cas9実験の際にはぜひ参考にしてください。
MISSION™ CRISPR ゲノム編集
ゲノム編集101 eBOOK
製品リストやご注文、お問い合わせ先などは以下のページに掲載しております。
<References>
*1 M Kosicki, et al. Prog Mol Biol Transl Sci. 2017;152:49-67.
*2 LA Gilbert, et al. Cell. 2014;159(3):647-61.
*3 AC Palmer, et al. Transcription. 2011;2(1):9-14.
*4 J Yang, et al. Stem Cell Reports. 2019;12(4):757-771.
*5 S Konermann, et al. Nature. 2015;517(7536):583-8.
*6 AC Komor, et al. Nature. 2016;533(7603):420-4.
*7 IB Hilton, et al. Nat Biotechnol. 2015;33(5):510-7.
*8 AV Anzalone, et al. Nature. 2019;576(7785):149-157.

下記フォームでは、M-hub(エムハブ)に対してのご意見、今後読んでみたい記事等のご要望を受け付けています。
メルクの各種キャンペーン、製品サポート、ご注文等に関するお問い合わせは下記リンク先にてお願いします。
*入力必須