これで完璧!DNA精製→PCR→電気泳動の実験フロー

細胞からDNAを抽出、精製し、PCRにかけて電気泳動で確認するという実験は、ライフサイエンスの研究室だけでなく、生物学科の学生実習でも頻繁に行われています。
そこで、初めてPCR実験を行う学生向けに、各ステップの原理やコツを解説します。PCRに慣れた経験者も、原理を把握しておくことで、応用やトラブルシューティングに役立つはずです。
ステップ①DNAを精製する
PCRに必要な材料のうち、多くのものは試薬として購入しますが、テンプレートとなるDNAだけは自分で準備する必要があります。DNAに不純物が混じっていると、PCR失敗の可能性が高くなります。また、DNA濃度が低いと、PCRで十分に増幅しません。
DNA精製において、一般的に行われる方法に、エタノール沈殿、RNase処理、フェノールクロロホルム抽出があります。
-
エタノール沈殿
研究室では「エタ沈」と省略して呼ぶことがあるほど、ライフサイエンスの研究では最もよく行われる操作の一つです。DNAは、水に溶ける性質がありますが、エタノールには溶けません。DNAを含む溶液にエタノールと一価陽イオン(材料は酢酸ナトリウム溶液など)を加えると、DNAが沈殿します。
エタノール沈殿は、精製だけでなく、沈殿させてから水を加えることで濃度調整ができるという利点もあります。
なお、エタノールを加えて-20℃で沈殿させるとき、DNA量が少ないサンプルでは、一晩保存すると回収率が向上します*1。 -
RNase処理
サンプル中にRNAが含まれていると、PCR時に非特異的産物が生じる原因になります。RNAを除去するため、RNaseを添加します。 -
フェノールクロロホルム抽出
タンパク質除去のためによく行われる方法が、フェノールクロロホルム抽出です。フェノールによってタンパク質が変性して不溶化し、クロロホルムを加えることで水層(DNAが含まれる溶液)にフェノールが混入することを防ぎます*2。
2層に分かれた溶液のうち片方だけを吸引する操作は、初心者には少し難しいかもしれません。事前に水などで練習するのもよいでしょう。
エタノール沈殿、RNase処理、フェノールクロロホルム抽出それぞれのプロトコールは、次の記事に書いてあります。ぜひ参考にしてください。
DNAサンプルをクリーンアップする方法
ステップ②PCRで任意のDNAを増幅する
DNAを精製したら、いよいよPCRです。
PCRの正式名称はpolymerase chain reactionで、日本語では「ポリメラーゼ連鎖反応」と訳されます。手元にあるDNAがわずかな量であっても、倍々ゲームの要領で、わずか数時間で理論上は約1億倍に増やすことができます。
PCRは、基礎研究に限らず、さまざまな場面で応用されています。お米や肉の品種の特定、遺伝子組換え作物かどうかの判定、さらには犯罪捜査や親子鑑定でよく聞く「DNA鑑定」にも、PCRが使われています。
<PCRの原理>
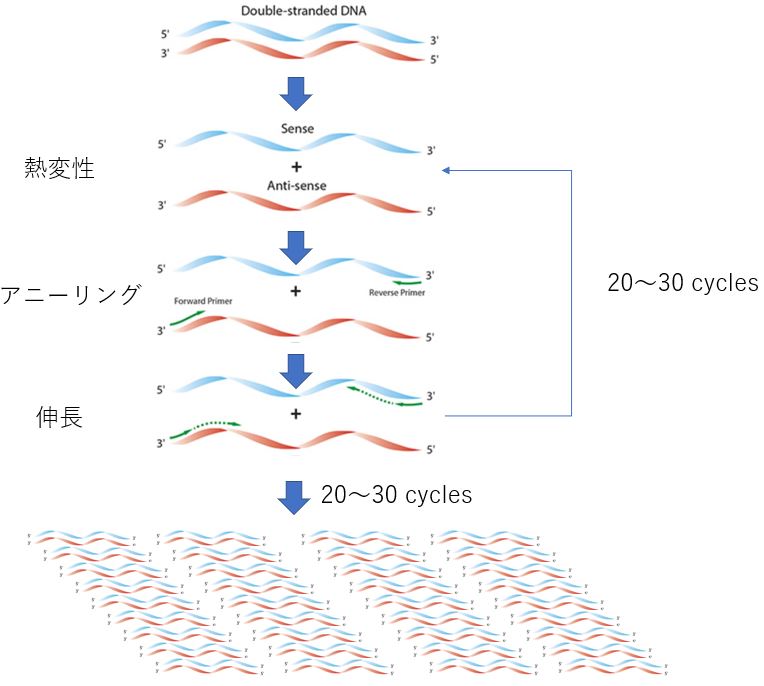
(出典:Polymerase Chain Reaction | PCR Process & Guide | Sigma-Aldrich、一部改変)
PCRは、DNAポリメラーゼによる細胞内のDNA合成を利用したものです。増幅させたいターゲットDNA配列の両端と相補的に結合できるオリゴヌクレオチド(プライマー)を用いて、増幅するターゲット配列を指定します。
PCRによるDNA増幅は、熱変性、アニーリング、伸長の3ステップを1サイクルとして、20〜30サイクル行います。
-
熱変性
90℃以上に加熱することで、2本鎖のDNAを1本鎖にし、プライマーが結合(アニーリング)できる状態にします。 -
アニーリング
温度を40〜65℃に下げると、テンプレートとなる長いDNAは1本鎖のままですが、20 〜30塩基からなる短いプライマーは、相補的な配列があるところと結合します。アニーリング温度は、プライマーの長さやGC含有率に依存します。 -
伸長
温度を約72℃に上げると、プライマーのあるDNA2本鎖の場所をDNAポリメラーゼが認識し、DNA合成が始まります。
<PCRに必要なもの>
-
PCR機器
PCRマシンやサーマルサイクラーとも呼ばれています。実験者が温度と時間、サイクル数をプログラムできます。 -
専用チューブまたはプレート
溶液中の温度が急速に変わるよう、200μLのように容量の小さいチューブを使います(いわゆる「8連チューブ」)。高スループットで実験をする場合は、チューブの代わりに専用プレートを使います。 -
テンプレートDNA
事前に精製し、指定した量(濃度)となるよう準備しておきます。 -
プライマー
論文など先行研究がある場合には、記載通りの配列を指定します。自分で設計する場合には、GC含量が40〜60%で、二次構造を作らないような配列になるよう考慮する必要があります*3。 -
DNAポリメラーゼ
一般的なDNAポリメラーゼではなく、高温に生息する細菌などからクローニングした専用の耐熱性ポリメラーゼを使います。
DNAポリメラーゼにも複数の種類があり、低温では不活性状態でサンプル調整中の非特異的増幅を防ぐ「ホットスタート法」に適したものや、エラー率の低さが特徴のものなどがあります。
目的に応じて最適なDNAポリメラーゼを選択すると、PCRおよびその後の実験の成功の確率が上がります。 -
dNTPs
DNA合成の際に取り込まれるdATP、dCTP、dGTP、dTTPの混合物です。
ここでワンポイントアドバイス。複数のテンプレートDNAに対して同じプライマーのセットでPCRを行う場合、テンプレートDNA以外の材料(マスターミックス)を本数分まとめて混ぜてから分注し、それぞれにテンプレートDNAを加えると作業効率が上がります。
そのとき、少し多めに作っておくと、「最後の1本分が足りない」という事態を回避できます。例えば、10本のチューブに分注する場合、マスターミックスは11本分作っておきましょう。
PCRの原理に関しては、以下に詳細が書かれています。
PCRとRT-PCRの基本原理
ステップ③電気泳動で任意のDNAを分離する
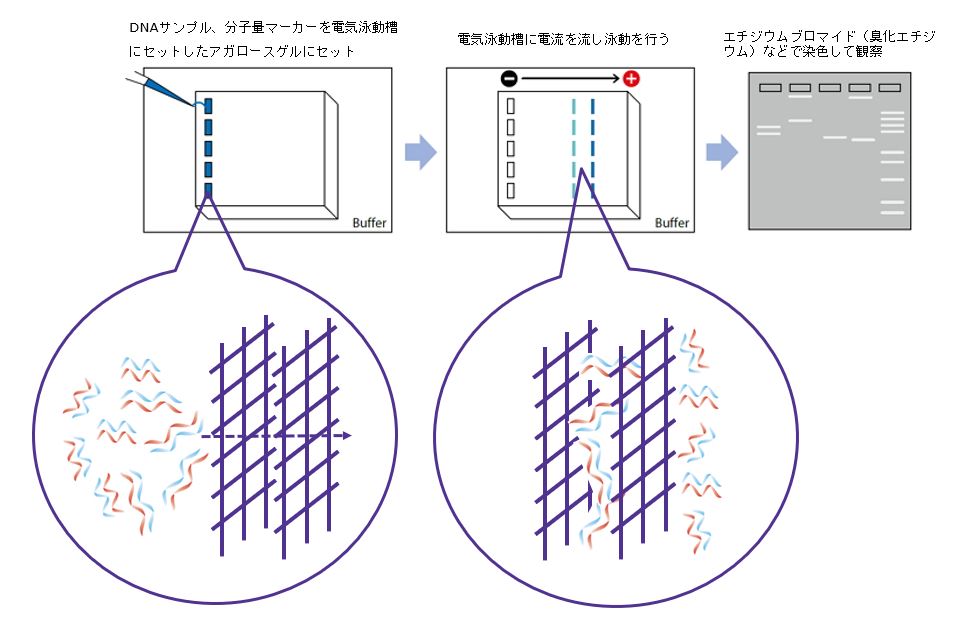
PCRを終えたら、目的のDNAが増幅したかどうか、アガロースゲル電気泳動でチェックします。
DNAは、リン酸基のところが負の電荷を帯びています。アガロースゲルで電流を流すと、DNAは陽極に引き寄せられます。
このとき、アガロースゲルの網目構造により、大きいものは網目構造に邪魔されて動きにくくなりますが、小さいものは比較的早く移動できます。
つまり、塩基配列が短いほど早く陽極に移動するため、長さでDNAを分離できます。
最後に、エチジウムブロマイドなどでDNAを染色すると、帯のようなものが見えます。これを「バンド」と呼びます。バンドの有無によって、DNAの有無を判断します。
電気泳動では通常、特定のサイズのDNAが複数混合されたもの(ラダーマーカーなどの分子量マーカー)を同時に流します。分子量マーカーとのバンドを比較することで、PCRで増幅したDNAの長さを推定でき、目的の配列かどうかの判断材料となります。
なお、アガロース濃度は、一般的には0.5〜2.0%で作成するのが基本です。アガロース濃度が高いほど、短いDNAを分離しやすくなります。
アガロース電気泳動を行ったDNAは、ゲルごと切り出して回収、精製し、クローニングなどに利用できます。
アガロースゲルの作成方法や電気泳動のプロトコールは、次の記事に書いてあります。
アガロースゲル電気泳動の原理と方法
原理がわかればトラブルシューティングに役立つ
DNAのPCRは、ライフサイエンスの研究室ならルーチンで行うほどありふれたものですが、基本を振り返ることでトラブルを解決できるヒントを得られるかもしれません。初心者だけでなく、PCRに慣れている人も、今一度原理を確認しておきましょう。
参照元
*1 春木 満「エタノール沈殿あれこれ」生物工学 第89巻(P.254より)
*2 NCGM脂質シグナリングプロジェクト・プロトコール集
*3 『PCR技術のすべて〜基礎から最新技術まで』(P.33より)


下記フォームでは、M-hub(エムハブ)に対してのご意見、今後読んでみたい記事等のご要望を受け付けています。
メルクの各種キャンペーン、製品サポート、ご注文等に関するお問い合わせは下記リンク先にてお願いします。
*入力必須