「逆合成解析」とは?基本的考え方から便利ソフトまでご紹介
複雑な化合物の構造を解析し、そのベストな合成ルートを合理的に立案する手法を「逆合成解析」と呼びます。本記事では、炭素の極性を基礎とした結合の切断、環形成の方法などの逆合成解析の基本的な考え方を、いくつかの実例を交えて紹介しています。
※無料トライアル実施中!逆合成解析の便利ソフトについて
逆合成解析とは何か
複雑な化合物の合成は、しばしば登山にたとえられます。頂上までのルートは無数に考えられますが、あちこちに危険な岩場や思わぬ障害があり、登山可能なルートはそう多くないものです。一見安全で簡単そうなルートに予期せぬ落とし穴があり、回り道に見えるルートの方が早く頂上に着けるようなことも少なくありません。
確実な登山のためには、事前の入念な調査が不可欠です。また、優れた道具を揃え、それらの使い方を熟知しておくことも重要になります。こうしてしっかりと計画を立てておけば、成功率はぐっと高まります。
有機合成では、この合成計画を「逆合成解析」と呼びます*1。ゴールとなる化合物の一段階前はこう、その前はこう……とさかのぼってゆき、入手可能な原料にたどり着くまでのルートを構築するものです。ルートはいくつも考えられますから、それらを比較検討し、最も効率のよいプランを選出します。
逆合成の考え方自体は古くから存在していましたが、これを集成・体系化したのはイライアス・コーリーです。彼はこの逆合成解析という方法論の開発を讃えられ、1990年のノーベル化学賞を単独受賞しています。
逆合成の基本
逆合成を考える際、炭素-ヘテロ原子結合の生成(たとえばカルボン酸からエステルへの変換)は一般に容易な工程であり、合成の最終段階付近で行うことが普通です。
また、アルコールからケトンやカルボン酸へ、あるいはその逆の変換(酸化段階の調整)も比較的容易で、合成の後期段階でも行いうる工程です。つまり合成計画を立案する上で、たとえば第二級アルコールとケトンは、「合成的に等価である」とみなすことができます。
一方、炭素-炭素結合の生成反応は、高温や強塩基を必要とするなど、反応条件が厳しいケースが多くあります。合成が進み、反応性が高い官能基が増えてから炭素-炭素結合を形成するのは、多くの場合難しいことです。
こうしたことから、複雑な化合物の合成においては、まず炭素骨格を構築しておき、そこに順次官能基を導入・変換するのが、基本的な流れとなります。
結合の切断
目標とする化合物の結合を(紙の上で)切断し、より単純で入手しやすい化合物へ分解していくことが、合成計画の第一歩となります。炭素-炭素結合の生成は、多くの場合、求核性炭素と求電子性炭素の反応によって起こりますので、切断した断片のどちらが求核性でどちら求電子性になるか、想定しながら切断していきます。
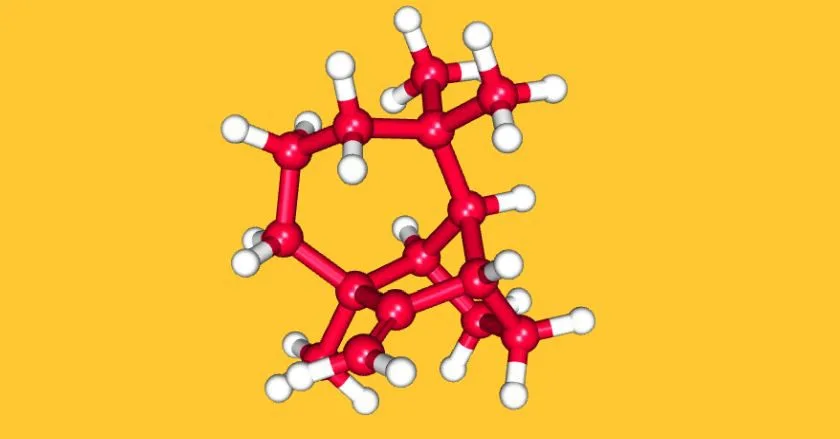
単純な例として、フェニル酢酸(1)を合成する場合を考えてみましょう。下の図の点線で結合を切断し、2つの断片(2, 3)に切り分けます。通常の化学反応は1本線の矢印で描くのに対し、逆合成の操作は白抜きの矢印で描き表すことになっています。
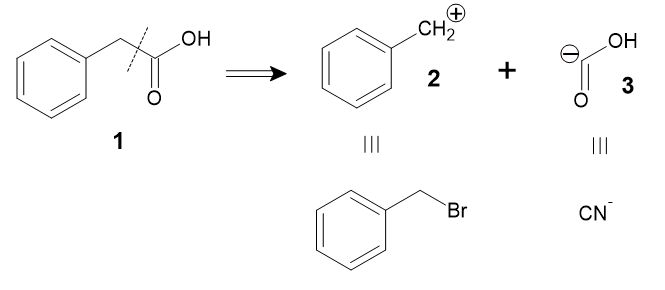
こうして得られる分子の断片を「シントン」(synthon)または「合成等価体」と呼びます。
といっても、たとえば-CO2Hというシントン(3)は、実際には存在できません。そこで、カルボン酸に容易に変換可能で、アニオンとして振る舞う化学種である、シアン化物イオンを用いることが考えられます。もう一方の断片であるベンジルカチオンは、実際には臭化ベンジルを用います。
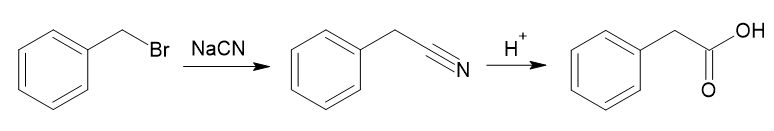
これをまとめると、臭化ベンジルとシアン化ナトリウムを反応させてフェニルアセトニトリルとし、これを酸で加水分解してフェニル酢酸を得るという計画が立てられます。
もちろん、他の切断の仕方もありえます。いくつかのルートを立案して比較検討し、最も条件に合ったルートを選ぶことになります。
官能基の活用
炭素骨格構築で鍵となるのが、目的化合物に含まれる酸素官能基です。特にカルボニル基は、Grignard試薬の付加やAldol反応など多くの炭素-炭素結合生成反応の足場となります。また、導入や除去も容易なので、逆合成解析における最重要の官能基といえます。
たとえば炭素鎖の1位と3位に酸素官能基がある場合には、Aldol反応を用いて結合を作ることが考えられます。Aldol反応は様々な立体制御法が開発されており、不斉炭素の構築が比較的容易なのも重要です。
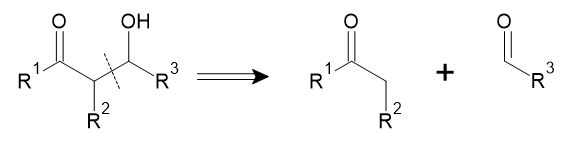
1位と5位に酸素官能基がある場合は、エノラートのMichael付加による方法が考えられます。このように、シントンの設計の際には炭素原子の極性に留意することが基本となります。
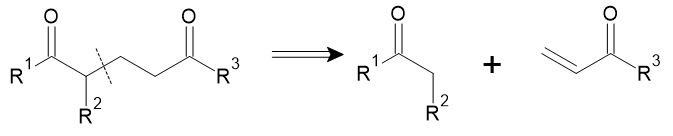
1, 2位に酸素官能基がある場合には、カルボニル炭素のアニオンに相当するシントンが必要になります。
この目的には、先ほど出てきたシアン化物イオンの他、1,3-ジチアンが利用可能です。イオウ原子に挟まれたメチレン水素は酸性が高まっており、強塩基を作用させるとアニオンが発生します。これをカルボニル化合物に求核付加させた後、ジチオアセタールを脱離させれば、1, 2位に酸素官能基が隣接した構造が得られます。
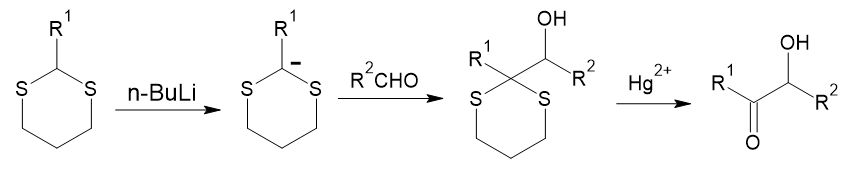
この一連の工程を通して見れば、通常求電子性であるカルボニル炭素を、求核性官能基に転換できたことになります。このように、シントンの極性を逆転させることを、「極性転換」または「Umpolung」と呼んでおり、逆合成における重要な概念です。
環状骨格構築
天然物の多くは、環状構造を持っています。中でも6員環構造を持つものは多く、その構築法は数多く工夫されています。
Diels-Alder反応は、一挙に多置換6員環を立体選択的に構築できるため、大変に有用です。分子間・分子内反応とも多くの利用例があり、6員環を含む化合物には、真っ先に適用を考えるべき反応です。
Robinson環化も、古典的な反応ながら今もよく用いられます。比較的入手容易な原料から、効率よく6員環骨格を得られる点で優秀です。また、芳香環をバーチ還元でシクロヘキサジエン骨格に導くのも、よく用いられます。特に、原料の芳香環は様々な置換基導入法が確立されていますので、検討に値する手法といえます。
7員環以上の中・大員環の環化はエントロピー的に不利であり、構築の難度は高くなります。しかし、オレフィンメタセシス反応が開発されて以降、これらの環の合成もかなり容易になりました。
特に各種のGrubbs触媒は官能基許容性が高く、環化のみならず炭素-炭素結合形成に広く適用可能です。逆合成の考え方を、最も大きく変えた反応といえるでしょう。
逆合成の実際
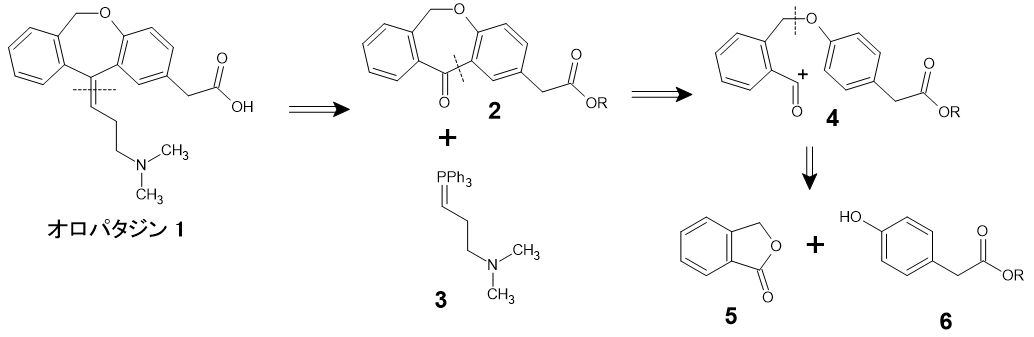
では、実際の逆合成の例を見てみましょう。抗ヒスタミン剤の一つ、オロパタジン(下図1)の合成経路を考えてみます*2。
まず目につくのは、分子中央の二重結合部分です。ここは、オレフィン形成の常套手段である、Wittig反応を用いることが考えられます。その原料となる7員環ケトン2は、分子内Friedel–Crafts反応で作る計画を立てます。さらにC-O結合のところで切断すれば、主要な原料4, 5, 6にたどり着きます。実際にも、基本的にこのルートでオロパタジンが合成されています。
もう少し複雑な例として、三環性骨格を持った天然物ロンギホレン(下図1)の例を考えてみましょう*3。コーリーらは、この骨格を点線のところで切断することを考えました。すると6員環と7員環が縮環した骨格となり、この分子の矢印のところを結べばロンギホレン骨格が得られることになります。
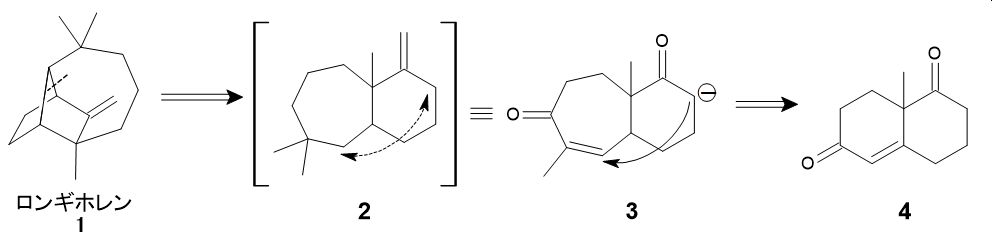
ただし、何の仕掛けもなしでは結合を作ることができませんので、3のような中間体を設定しました。ここに強塩基を作用させてエノラートとし、分子内Michael付加を行えば、ロンギホレンの骨格が完成するという設計です。そして中間体3は、二環性ケトン4に環拡大反応を施すことで得られると考えられます。4はWieland-Miescherケトンと呼ばれ、容易に合成可能な中間体です。
ロンギホレンは他にも多くの合成例があり、様々なアプローチが行われています。逆合成のエッセンスを学ぶための、よい題材といえるでしょう。
逆合成解析はコンピュータとの相性もよく、初期から検討されていました。近年ではさらに精緻になっており、たとえば「SYNTHIA®」は、化学者の経験知やネガティブデータなどを取り込んだことで多くの合成ルートの立案が可能となった、極めて頼りになるツールです。
とはいえ、コストや難易度なども考慮して、どのルートが一番よいか判断を下すのは結局人間です。優れた合成の論文を数多く読み込み、善し悪しを判断できる目を養うことが、何より重要と思います。
逆合成解析をより身近に―SYNTHIA® Lab―
有機合成化学とコンピューター科学を組み合わせることで開発された逆合成解析ソフトウェア「SYNTHIA®」に、アカデミアのお客様向けにご利用しやすい新プラン「SYNTHIA ® Lab」が登場しました。
無料トライアルもご用意しております。ご登録いただき、ぜひお試しください。
SYNTHIA® Labの詳細についてはこちらをご参照ください
逆合成解析ソフトウェアSYNTHIA®に関するお問い合わせはこちら
<References>
*1 「コーリー 有機化学のコンセプト」 E. J. Corey, Cheng Xue-Min著 丸岡啓二訳 丸善出版
*2 「創薬化学 有機合成からのアプローチ」 北泰行・平間哲夫編 東京化学同人 p.291
*3 E. J. Corey et al. J. Am. Chem. Soc. 86, 478 (1964).


下記フォームでは、M-hub(エムハブ)に対してのご意見、今後読んでみたい記事等のご要望を受け付けています。
メルクの各種キャンペーン、製品サポート、ご注文等に関するお問い合わせは下記リンク先にてお願いします。
*入力必須